Using Celery to scale bioinformatics analysis
Celery is an asynchronous task queue/job queue based on distributed message passing
Yes I know, there are tons of tutorials on how to run Celery out there, but I just wanted to showcase how we use it in our production environment, this is a real life example.
What is celery? How does it work?
Celery is an asynchronous task queue/job queue based on distributed messaging passing. Plainly speaking, and taking out complexities, what this means is that you will have a queue of messages produced by someone that we will call producers. Then you will have someone, we will call them workers, reading this messages and doing some work. The following picture would represent this workflow:
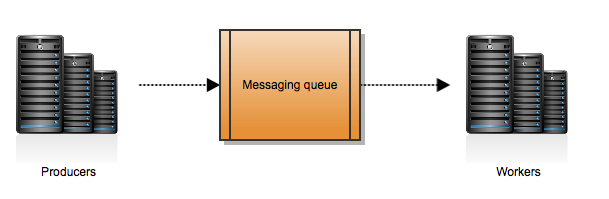
This is the most basic Celery architecture you can have. Celery can work with several messaging queue systems, called brokers. We use RabbitMQ for our production environment. But you can use several.
As I said at the beginning, there are tons of tutorials out there, being the official one very good. So I will skip the “How-To” and just describe our environment.
How do we use Celery?
Bcbio-nextgen is the genomics pipeline we use at Science For Life Laboratory for the analysis of our samples. It is based on Python and developed by Brad Chapman. Celery is also written in Python, so the integration of the pipeline tasks with celery is straightforward.
A basic analysis would go as follows:
-
The raw data that comes from the sequencing machines (BCL image files) needs to be converted to something we can work with, i.e FASTQ files. At the same time, a demultiplexing process of the samples is carried on by Illumina software.
-
Once this is done, several steps compose an analysis, that is done using this FASTQ representation of the sequenced samples. These steps can be atomic, though we try to run the whole analysis in a row. Summarizing, the steps of a complete basic analysis would be: Sequence alignment, contaminants removal, merge samples, mark duplicates and variant calling.
NOTE: Sorry if you are not familiar with Gemone Sequence Analysis, but the important thing to note here is that these tasks can be atomic.
Step one is done locally on our processing servers because it is very I/O intensive and our disks are fast. But step two is very CPU/memory intensive, and we need to do the analysis somewhere else. Here is where Celery comes in play. We are using an HPC center where we have our workers. These workers are listening to different message queues. When the preprocessing finishes on our servers (and the necessary data is sent to the HPC), these servers send a message “analyze” to one of this queues. When the workers pick up this message, the complete analysis starts. This figure illustrates our architecture:
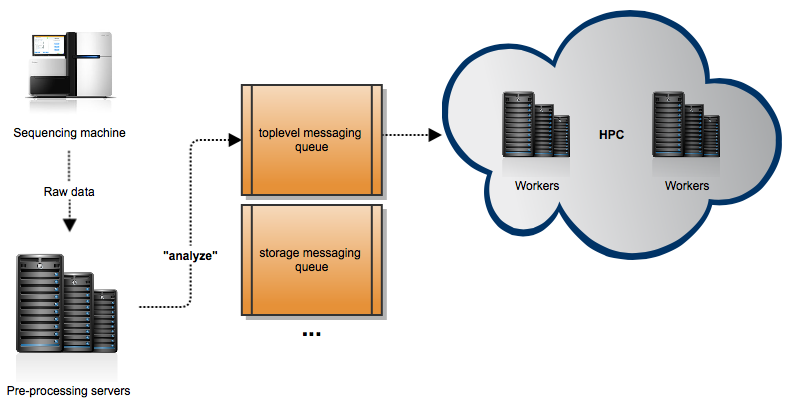
You may think that we are not properly using Celery, as even if the analysis can be split in several steps, we are sending a message that basically says: Do… everything! Well, this is partly true. Actually the pipeline is designed to be able to restart the analysis at any point, is just that we almost never have to do that. Take a look at all the tasks that we have defined. Also, the main task “analyze” basically launches a program that takes care of atomically send jobs to SLURM, the queueing system used in our HPC.
The benefit of using Celery is that it can Scale to as many Workers as you want. Each worker will pick up a task and return the result if required, all of this asynchronously (or polling if you want to). Have in mind though that workers and producers should scale evenly. Here there is a very good presentation by Nicolas Grasset of things you should consider when working with Celery.
I hope that you enjoyed the reading, the intention of this blog post was, as I said, just to show a practical example of how Celery can be used.
