Genomics reference data: The fragmentation problem (part II)
This blog post is the continuation of “Genomics reference data: The fragmentation problem (part I)”.
In part I, we saw that the majority of the research groups work with several species and they need to download lots of reference data. We also saw that the sources for this data are diverse and unstructured. With the remaining questions I try to know how people is fetching and structuring reference data.
Questions 4, 5 and 6: How do you fetch your reference data? How do you structure your reference data? How do you keep your reference data up to date?
I decided to group these questions because they’re so closely related: You fetch your reference data and store it somehow. Later, from every now and then you have to update the reference data. If you fetch data from several places and how you get it (the structure) is different for every place, you’ll probably end up creating your on structure.
Here, some results:
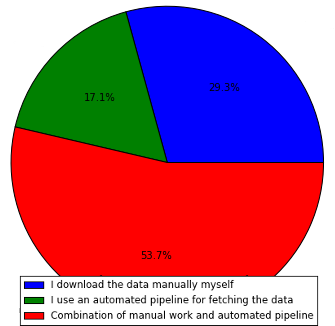
As expected, the majority of the people uses a combination of automated and manual work. Quite surprising the amount of people that downloads the data manually themselves, with a 29.3%. If we sum up, a total of 82.9% of people do some kind of manual work for something that, IMHO, should be completely automatic. We are of course in the red group as well.
One particularity is that, if we group the answers by “Do you work only with one species, or several species’ genomes?” and “How do you fetch the reference data?”, we can see that only 2/7 of the people that answered “Only one specie” answered as well “I use an automated pipeline for fetching the data”. I would expect the opposite relation here, as fetching data for only one specie should be easier to automate.
How do you structure the reference data?
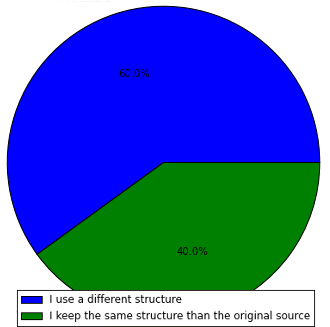
I am quite surprised with this result actually. I honestly thought that a higher percentage of people would use a different structure, given the amount of people that fetches reference data from different places and species. At least that is what we try to do: To unify everything under the same directory tree, which implies merging datasets from different places in a single place.
I also asked the question “What motivates you to use your own structure for your reference data?”. This was an open-ish question and thus difficult to plot, so I’ll just paste here the relevant answers. Again remember that you can obtain the responses form in my GitHub repository for this blog:
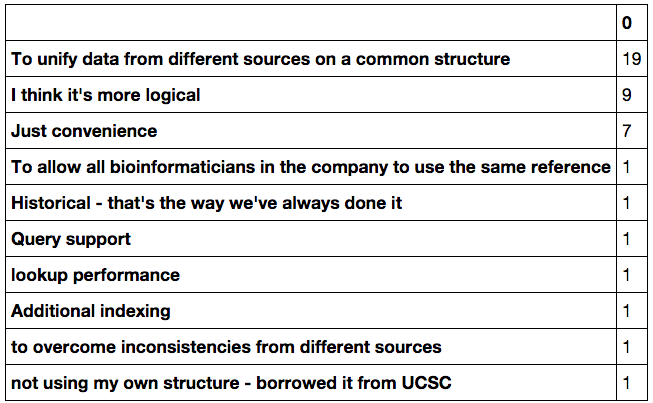
Indeed the major reason for structuring the data on a particular way is unification and convenience. The “I think its more logical” answers evidences the fact that in some particular sources of reference data, you can be very lost at the beginning….
Question 7: How do you keep your reference data up to date?_
This is also interesting, and the answers to this questions were quite disappointing, in the sense that there doesn’t seem to be any way to do this nicely… Again I will just paste here the set of answers:
- check at start of new projects
- I infrequently check for new updates and fetch them manually
- do manual check : Most of the time we stick to stable version e.g. we are still using hg19
- I don't
- regular downloads quarterly
- custom cron scripts
- I use one of the aforementioned tools to automatically check for new versions of the reference data
- custom tool
- following the corresponding mailing lists
- Generally only want to use one version for a given project. Check for new versions if applicable when starting something new.
- cron+wget/rsync for most sources
- I regularly check for new updates and fetch them manually
- when i remember or someone needs a newer version
- prefer working with one version across the project
- only when need
- I update data when I start a new project
- Staying consistent with versions of references is more important than having the latest references.
- Update as projects demand
As you can see, the responses are quite diverse, and it involves a lot of manual work. Lots of answers are on the lines “when a project starts” or “on demand”, again making evident that the difficulty on automation of this process makes it hard to catch up with.
Question 8: Do you use any of these tools for downloading reference data?_
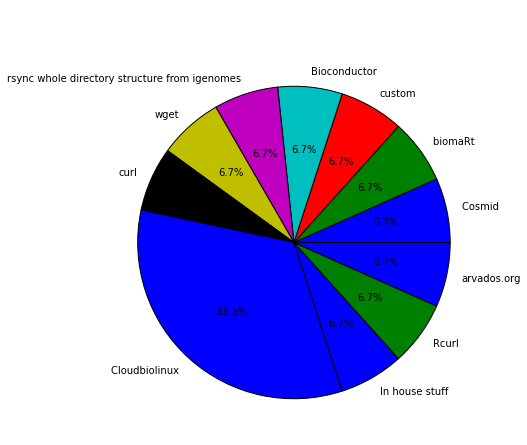
There are lots of tools to help with the task of fetching reference data. Cloudbiolinux seems to be the most used among the participants, with 5 users. Whilst for the rest of the tools, apparently everyone uses its own. NOTE: There were lots of empty answers to this question, so if you make the numbers it won’t sum the total number of participants.
Cloudbiolinux is indeed a very complete tool that helps a lot in fetching reference data. We use it all the time, but still there are some types of data or organisms that are not available there.
It would be so nice to have a single tool for this task…
Conclusions
It is clear that something is wrong with the organization of genomics reference data. As I said on several occasions in this post, should be fairly easy to download and update such an important part of a genomics analysis.
The problem seems to be that organizations need to fetch data from several places and then structure it in some way adapted to their needs. Also, the data is available only in some particular servers (UCSC, ENSEMBL, etc) so people have to adapt to them. This is a problem for convenience, availability and reliability: What happens if a service is down? or if they decide to change the data structure? Something needs to be done here…
BioMart seems to be a good approach. Federation of the data would be a good win for both convenience and reliability. But we still have the problem of availability: It is the labs who host the data, so in case someone decides to stop hosting the data, no one can access it anymore again. Unless, of course, someone else has previously downloaded it and wants to publish it again in BioMart. Wouldn’t it be fantastic if that distribution was automatic? This scenario reminds me quite a lot to the BitTorrent protocol, doesn’t it? That’s what me and Roman Valls think, at least.
- Someone (UCSC, ENSEMBL, etc) publishes some reference data, and starts distributing it through this protocol.
- Inherently to the protocol, the data starts distributing all across the clients that download it, becoming more available and reliable.
A top layer could be added to be able to create logical or custom directory structures, still pointing to the same torrent files, so that users can download data and store it already in a particular structure, if desired. Also one could implement another layer to automatically build indexes for several aligners using some kind of virtualization like Docker, for example.
Of course this is just an idea and would need a lot of work on designing and thinking first, but I would love to know your thoughts about something like this, or about the situation in general. Also one needs to have in mind that BitTorrent is a stigmatized protocol in HPCs, so would have to deal with that as well.
And this is all for the survey. I must apologise again because the format of the survey (totally my fault) made it quite difficult to parse and extract conclusions, but as I said in Part I, one learns from this kind of mistakes :-)
Please share if you found this post useful, and feel free to comment!
